Cloning
The objective of molecular cloning procedures is to introduce a gene of interest into a suitable expression host in a compatible manner (recombinant DNA) to ultimately produce the gene product (protein) in satisfying quality and quantity. Cloning means producing identical copies of a certain gene of interest. By cloning, the gene is amplified and then inserted into a plasmid (vector) for the following replication and protein expression. Using plasmids allows for the protection of the foreign genetic material from the expression host’s innate degradation machinery.

There are different cloning procedures used to insert the gene of interest into a plasmid. Restriction enzyme-based cloning is the standard cloning method in molecular biology. Restriction endonucleases cleave double-stranded DNA (dsDNA) at specific sequence sites called recognition sites. Depending on the employed restriction enzyme, the generated DNA fragments can either have blunt ends or sticky ends. These can be fused with plasmid DNA if it is cleaved with the same restriction enzyme and thus linearized. The fusion of the DNA fragment and the linearized plasmid DNA takes place with the help of a so-called DNA ligase.
A special restriction enzyme-based cloning method makes use of the specific properties of type IIS restriction enzymes, endonucleases that cleave DNA outside of the recognition sequence. In contrast to traditional restriction enzyme cloning, this cutting outside from the recognition site provides the advantage that custom overhangs can be generated. With the StarGate system, IBA has utilized this approach to create compatible custom overhangs, which can then be fused efficiently. The StarGate system comes along with a large subset of cloning and expression vectors for E. coli or mammalian cells.
StarGate for fast protein expression cloning
StarGate has been developed for the rapid systematic screen of the optimal expression (regarding expression host and/or promoter) and purification (regarding the fusion tag) system for a given gene of interest (GOI).
There are two possible cloning procedures.
Direct Transfer Cloning

The Direct Transfer Cloning procedure is recommended in cases the optimal expression and purification system is already known. In this procedure, a PCR product containing the gene of interest can be directly inserted into the appropriate Expression vector (Acceptor Vector).
In the first step, the gene of interest (GOI) will be equipped at both termini with combinatorial sites (orange and red) and the Esp3I recognition sites (light blue), which are important for oriented insertion of the PCR fragment. This PCR product is in the second step integrated into the appropriate Expression vector resulting in the final expression construct, the so called Destination Vector. The formation of the correct Destination Vector is monitored via blue-white screening on LB-Agar plates containing X-Gal. used for transformation or transfection of the expression host.
“Two-step-cloning” via pENTRY-IBA
StarGate cloning via the pENTRY-IBA vector provides a tool for the systematic and rapid screen of the best working expression conditions for a certain GOI, where nothing is known from literature. After initial pENTRY cloning, the GOI in the resulting Donor Vector can be easily transferred by a simple one-tube reaction into a multitude of Acceptor Vectors providing different features.

Step 1: Entry Cloning for Donor Vector generation
In the first step, the gene of interest (GOI) will be equipped at both termini with combinatorial sites and the LguI recognition sites, which are important for oriented insertion of the PCR fragment into pENTRY-IBA51. This is done by PCR using a proofreading polymerase.
Recombination of the PCR product with the Entry Vector at the combinatorial sites (red and orange) leads to generation of the Donor Vector. This step involves loss of the LguI restriction sites (dark orange with arrowheads), making the recombination reaction unidirectional and thereby highly efficient.
In the resulting Donor Vector the same combinatorial sites are now flanked by the Esp3I recognition sites (light blue), thereby enabling a highly efficient and specific StarGate® gene transfer process into Acceptor Vectors in a similar manner.
")
Step 2: Transfer Cloning (Destination Vector Generation)
In the transfer reaction step, the GOI is transferred from the Donor Vector into an Expression vector (Acceptor Vector). The Acceptor Vectors provide the desired genetic surroundings (i.e. affinity tag, promoter (prom), additional signal sequences, etc.). In cases the optimal expression and purification conditions of the GOI are unknown, a variety of different Acceptor Vectors should be chosen.
The transfer from the Donor Vector into the Acceptor Vector is assisted by Esp3I as restriction enzyme and T4 DNA Ligase. In the resulting Destination Vector, the GOI is placed under control of the selected promoter (prom), allowing GOI expression in the selected expression host. In this example, an affinity tag (green) is fused to the C-terminal end of the GOI expression product.
Special features provided by StarGate Cloning
Cloning via Gene Synthesis
Gene synthesis – the easy way to generate your StarGate Donor Vector

How to generate a StarGate Donor Vector by using a gene synthesis service:
- Enter the sequence of your GOI without start and stop codon
- Select specific codon optimization for your final host. Add the Esp3I recognition site and the respective combinatorial sites:
Start: 5'-AGCGCGTCTCCAATG
Stop: 3'-GGGAGGAGACGCGCT - Choose a standard cloning vector (offered by the service provider) containing an antibiotic resistance gene for kanamycin
Expression Vectors (Acceptor Vector)
IBA Acceptor Vectors provide different genetic surroundings for optimal protein expression and purification. These include different purification tags, promoter for a variety of host cells, signal sequences etc.
E.Coli
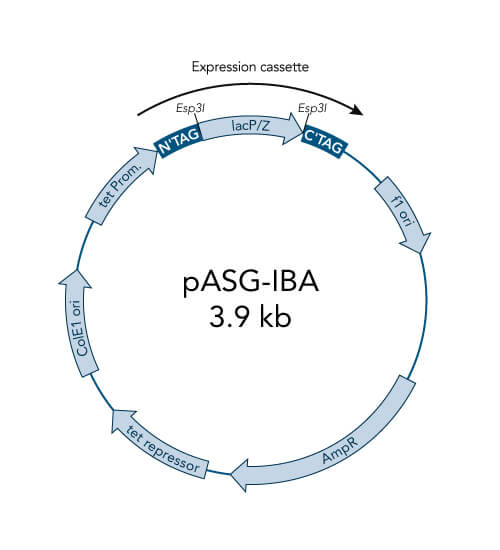
pASG-IBA vectors
- High-level expression in E. coli, also for toxic proteins
- Tightly regulated expression due to anhydrotetracycline (AHT) inducible tetA promoter/operator
- Option for periplasmic expression due to ompA signal sequence
- No catabolite repression - no influence of medium components
- Not influenced by the genetic background – wide choice of E. coli expression strains
- Inexpensive induction with AHT
Mammalia
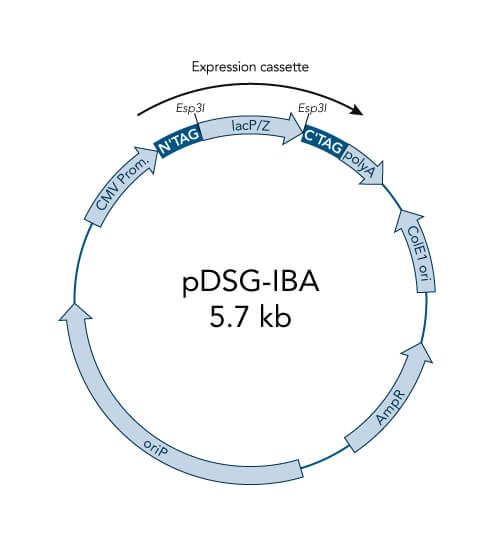
pDSG-IBA vectors
- Optimized for MEXi, IBA´s mammalian expression system, and are also suitable for other mammalian cell cultures
- High level constitutive expression in mammalian cells by the CMV promoter
- Extrachromosomal replication due to Epstein Barr Virus replication origin (oriP) - requires chromosomal expression of the EBNA-1 gene, e.g. like in MEXi 293E cells
- ampicillin resistance cassette for selection of transformed E. coli cells
- ColE1 origin for a high plasmid copy number
- BM40 option for secretion of protein into the medium
Mammalia
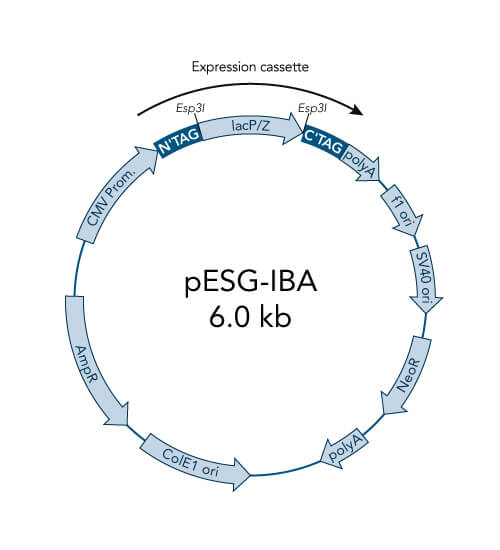
pESG-IBA vectors
- High-level constitutive expression in mammalian cells by the CMV promoter
- Neomycin resistance for generation of stable cell lines
- ampicillin resistance cassette for selection of transformed E. coli cells
- ColE1 origin for a high plasmid copy number
- BM40 option for secretion of protein into the medium
Mammalia
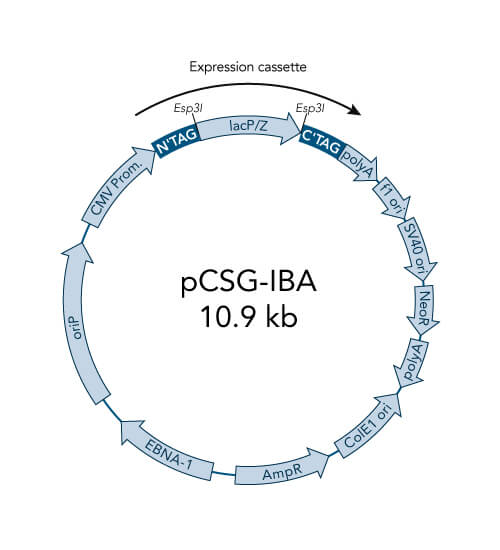
pCSG-IBA vectors
- High-level constitutive expression in mammalian cells by the CMV promoter
- extrachromosomal replication due to Epstein Barr Virus replication origin (oriP) and
- chromosomal expression of the EBNA-1 gene is not required, because the EBNA-1 is encoded by the vector
- prolonged expression of the inserted GOI under G418 selection without the need for making stable cell lines
- ampicillin resistance cassette for selection of transformed E. coli cells
- ColE1 origin for a high plasmid copy number
- BM40 option for secretion of protein into the medium
Tet expression system

Features and benefits of the pASK-IBA vectors:
- High-level expression in E. coli
- Tightly regulated expression due to the tetracycline promoter
- Enhanced stability of cytotoxic genes
- Inexpensive induction with anhydrotetracycline
The pASK-IBA vectors are available with Strep-tag®II, the OmpA secretion signal, special protease cleavage sites and Chloramphenicol resistance.
Principle and properties
pASK-IBA vectors contain the strictly regulated tetracycline (tetA) promoter, which is suppressed by the tet repressor until induction with a low concentration of anhydrotetracycline. This prevents premature expression of the foreign gene. The tet repressor is also encoded on the pASK-IBA vector and is constitutively expressed by the chloramphenicol acetyltransferase promoter. However, the vectors do not confer resistance to tetracycline.
Unlike the T7 promoter, no special E. coli strains or additional plasmids are required. In contrast to the lac promoter, the tetA promoter/operator is not prematurely expressed without an inducer and is not functionally linked to cellular regulatory mechanisms or a specific genetic background. Furthermore, compared to the lac promoter, the tetA promoter is not susceptible to catabolite repression (cAMP level, metabolic state) and is not influenced by chromosomally encoded repressor molecules.
As a consequence, special E. coli strains or extra plasmids are not required and a broad range of culture media and conditions can be used. For example, glucose minimal media and even the XL1-Blue bacterial strain, which carries an episomal copy of the tetracycline resistance gene, can be used for expression. The pASK-IBA expression system is stable under many conditions, including fermentation, and is easy-to-handle.
Further elements of the vectors are a tandem ribosome binding site (RBS) which ensures efficient initiation of translation, the strong terminator of the lipoprotein gene in order to prevent read-through, the intergenic region of the bacteriophage f1 which provides a means for preparing ssDNA and a chloramphenicol acetyl transferase gene. The vectors do not mediate resistance against tetracycline.
Reference: Skerra, A. (1994). Use of the tetracycline promoter for the tightly regulated production of a murine antibody fragment in Escherichia coli. Gene, 151, 131-135.
The following E. coli strains have already been used successfully for Tet expression with our pASK-IBA vectors:
- JM83
- WK6
- B
- BL21
- MG1655
- W3110
- BL21(DE3)
- BLR(DE3)
- XL1-Blue
- BL21-CodonPlusTM-RIL
For secretion, we recommend JM83. For cytoplasmic expression E. coli B strains are recommended, since they lack the lon protease and the ompT outer membrane protease that can degrade proteins during purification (Grodberg and Dunn, 1988, J.Bacteriol. 170, 1245).
Please note that we are not aware of an E. coli strain that is incompatible with the Tet expression system.
Anhydrotetracycline: Inducer for tetA Promoter
The expression cassette of pASK-IBA vectors is under transcriptional control of the tetA promoter/operator and repressor. The promoter is induced by a low concentration of anhydrotetracycline (AHT) saving costs and minimizing the antibacterial influence of AHT. Degenkolb et al. (1991) have shown, that AHT binds 35-times tighter than tetracycline to the tet repressor.
TypeIIS Restriction Enzymes
Type II enzymes are one of the 4 (I-IV) types of recognized endonucleases, which cut DNA at a particular recognition site. Type II enzymes cleave within or a short distance from their recognition sites, which comprise usually 4–8 nucleotides in length.
Among them, are the type IIS enzymes, like LguI and Esp3I.
Type IIS restriction enzymes are dimeric enzymes that cleave DNA at a defined distance from their non-palindromic, asymmetric recognition site. This means that the target sequence can only be read in one direction. Thereby the digestion with only one single enzyme can generate two different independent sticky ends with 5’-overhangs allowing directional cloning. In addition, after digestion reaction the recognition sequence is removed completely and therefore the encoded amino acid sequence is not affected by remaining restriction enzyme sites.

The usage of typeIIS restriction enzymes provides important features for cloning:
- It allows one tube cloning
- Expression of authentic proteins is possible (no additional amino acids)
- The cloning will be always in frame with the vector features
- Assembly of multiple fragments is possible
References:
1. Pingoud A, Jeltsch A (2001). Structure and function of type II restriction endonucleases. Nucleic Acids Res. 29 (18): 3705–27.
Application example
StarGate comprises vectors for expression in mammalian cells (pCSG-IBA, pESG-IBA, pDSG-IBA) and E. coli (pASG-IBA). As an example, the bacterial protein azurin was cloned into different pASG-IBA vectors and expressed in E. coli. Expression results were analyzed by SDS-PAGE.

The coding sequence of azurin, a 14 kDa bacterial protein, was cloned into 10 different pASG-IBA vectors and expressed in E. coli. Comparable amounts of E. coli cells were harvested 3 hours after induction with anhydrotetracycline. Samples were lysed by boiling for 5 min at 95 °C with gel loading buffer and analyzed by SDS-PAGE. Subsequently, the gel was stained with Coomassie. Periplasmic secretion by means of ompA led in all cases to accumulation of comparable amounts of the protein of interest (lanes 6-10). This could be expected as azurin is also secreted in its authentic host P. aeruginosa. In case of cytosolic expression, however, interesting aspects became obvious, since it was not anticipated that cytosolic expression of azurin is possible at all. Expression was enabled by fusion of an N-terminal affinity tag (lanes 2, 4, and 5), while N-terminal untagged variants did not result in expression (lane 1 and 3). The examples show that initial screening for expression conditions may be worthwhile since different constructs can lead to various results depending on the target protein properties.
